广州供卵的价格一般多少,广州找代妈生孩子多少钱,

医百病导读:
有人问我,如果看不到确定的未来,还要不要付出。我只能说,并不是每一种付出都是在追寻结果。有时在付出的路上,能够收获的,是清楚地看到了自己想要的,或者不想要的,这又何尝不是一种宝贵的结果。命运会厚待温柔多情的人,好过冷漠的一颗心
文丨世界那么大还事遇到你
三岁的她就患上了“老年痴呆”?
|她开始忘了爬行、坐立.
·HollyChandler在20个月大时被诊断出患有C型尼曼-匹克病
·这种攻击大脑的病症,在英国的患病率为1/120000
·医生们透露,来自达德利的Holly已经失去保持站立的能力
·为了全职照护女儿,母亲ElaineFisher辞去了店员工作
一个突然忘记如何爬行、坐起和站立的“健康”学步孩童,被诊断出患有类似于阿尔茨海默病的疾病。
3岁的HollyChandler在20个月大时被诊断出患有C型尼曼-匹克病(Niemann-PickTypeC,NP-C),这种病会以类似于阿尔茨海默病的方式攻击她的大脑。
38岁的单身母亲ElaineFisher来自西米德兰兹郡达德利,在女儿令人心碎的诊断后,一直处于“哀伤”之中。
医生们透露,由于这种罕见病,Holly已经失去保持直立的能力。英国大约每12万人中有一人遭受该病的侵袭。
Fisher女士还有3个儿子,在Holly一岁生日时,她首次注意到女儿在一次病毒感染后出现了一些问题。
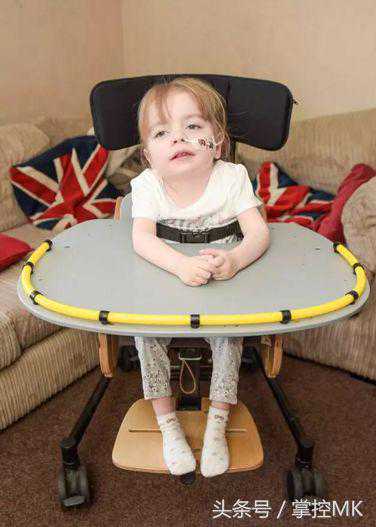
Fisher女士把Holly和自己同龄的孙子做了比较,意识到Holly停止了生长发育的里程碑过程,并开始忘记之前已经达到的那些。
在只比Holly大一周的孙子Jacob已经开始走路和更多互动时,Holly却在退步。
广州夫妻不能生找代妈住院9天接受大量检测后,Holly获得诊断。Fisher女士现在下定决心要尽可能给女儿最好的生活质量。
Fisher女士放弃了自己的店员工作,成为Holly的全职看护人,她说:“我不得不为我将永远无法拥有的女儿哀伤,同时会尽我所能地学会照顾Holly。”
“我一直在想所有我们能够一起做的事,就像我和妈妈一起做的那样,购物和外出,但这已经被剥夺了。
“要理解这种可怕病症是很不容易的,最好是把它想成阿尔茨海默病,因为它们以相同的方式攻击大脑。
“她仍然是一个可爱、活泼、有趣的小女孩,她喜欢叮当小仙女(Tinkerbell)。她有大堆的小叮当玩具。但她已经开始倒退了。
她补充道:“她曾经能够站立、坐起和爬行,但现在不能了,因为她大脑的那些部分受到了攻击。
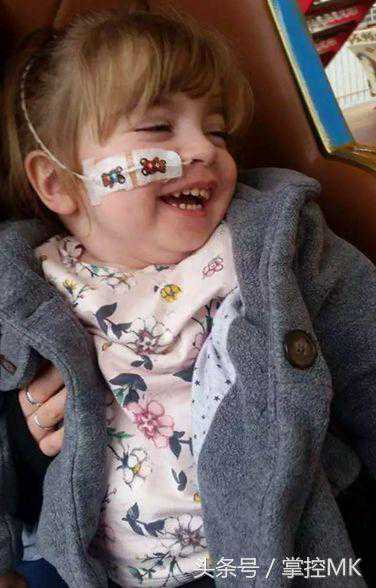
“有时她只是躺在地板上,什么也不做,甚至都没有眼神接触,尽管她现在更善于交流。
“我们不确定和她在一起的时间还有多久,没人能告诉我们。
“她可能活到20多岁,但也可能只有两三年时间。所以我们要确保能留下所有的记忆。
——Fisher女士的妊娠——
2014年10月,医生们发现胎儿不再生长后,Fisher女士在怀孕37周时通过剖腹产生下了Holly。
不过,Holly出生时体重5磅,看起来很健康,所以医生第二天就让她们出院了。
Fisher女士说:“怀孕过程很正常。我早上孕吐很厉害,只想吃甜食。
“怀孕的最后一个月,他们开始监测我的状态,因为Holly已经停止生长。他们决定进行剖腹产,然后Holly就出生了。
“她很小,只有差不多我两只手那么大,大约6周时穿新生儿衣服依然显大。”
——注意到有问题——
广州国内供卵中心Holly继续正常发育,学会了爬行、坐起,并能够拉着东西站立。
但在她一岁生日后不久,Fisher女士注意到有些不对劲。
在又一次感染鹅口疮之后,Holly没有恢复正常。
Fisher女士的孙子和Holly年龄相仿。当Holly的生长发育开始落后于她的孙子时,她知道出了问题。

Fisher女士说:“在12个月大时的检查中,她很好,没有任何问题。然后差不多一岁生日时得了鹅口疮,就没有恢复正常。
“她看起来很疲劳,不吃东西,而且不再是那个我认识的正常、活泼的小女孩。
“我有一个比Holly大一周的孙子,他当时能够走路,而Holly却不行。
“看到他们在一起的样子,你就会发现有些不对劲。”
2016年4月,Holly因耳朵和喉咙感染被达德利拉塞尔霍尔医院收治。
她在医院住了9天,其间医生们注意到她的脾脏肿大并开始进行检查。
——诊断患有C型尼曼-匹克病——
Holly被转诊到伯明翰儿童医院,医务人员在她的脾脏中发现了脂肪沉积物,表明是C型尼曼-匹克病。
Fisher说:“我知道这很严重,因为他们告知我的时候,房间里大约有6名工作人员。
“他们询问我她的状况,然后解释说她患有C型尼曼-匹克病。我从未听说过这种病,一时反应不过来。
“然后医生说这是一种会攻击她大脑的疾病,以至于将大脑完全破坏。我的世界顿时崩溃了,我记得泪水夺眶而出,但不记得医生们说了什么。”

C型尼曼-匹克病会导致一些患者在儿童早期出现症状,而另一些患者则可能多年无症状。该病会导致脂肪或脂类在肝脏、大脑和脾脏中积累,破坏细胞。当大脑受到该病攻击时,则会引起痴呆样症状。
Fisher女士说:“把这种疾病描述为阿尔茨海默病是准确的,当我需要向别人简单解释时,我经常这样描述。
“因为阿尔茨海默病攻击大脑的方式与C型尼曼-匹克病相同。”
——专注于生活质量——
Fisher女士患有克罗恩病,但她现在要专注于努力给予Holly可能的最佳生活质量。
她晚上通过鼻饲管进食以保持体重,并接受药物治疗以防止疾病和其他感染。
Fisher女士和她的3个儿子(20岁的Owen、17岁的Josh和16岁的Alex),在家里照顾Holly。
Holly还会每周两次去一个配有专职护理员的托儿所,这是她喜欢的。
Fisher女士说:“我们尽可能多地出去玩,拍很多照片,这样我们就能拥有回忆。不过我们必须避免去吵闹的地方,因为Holly不喜欢。
“她喜欢玩耍,会把她的手放在我的手上,然后指向她想要的东西。我们要确保她尽可能保持快乐,并且喜欢和我们在一起。
“她是我的宝贝小女儿,我不知道没有她我该怎么办。”
C型尼曼-匹克病是什么?
C型尼曼-皮克病(NP-C)是一种各年龄段均可发病的罕见遗传性神经退行性疾病。发病年龄可有很大差异,大约50%的病例见于儿童期。NP-C是由肝脏、大脑和脾脏的脂类或脂肪累积引起的。症状包括脾脏或肝脏肿大、行走困难、四肢姿势困难、言语不清、学习困难和进行性智力衰退。当缺陷基因(突变)的两个拷贝传递给孩子时,孩子就会遗传该病。
如果夫妻双方都携带缺陷尼曼-匹克基因的一个拷贝,每次怀孕,他们的孩子都有25%的几率罹患NP-C。据估计,每12万人中有一人患有NP-C,不过最近的证据表明这可能被低估。NP-C目前无法治愈,但是患者可以受益于缓和疗法。
目前正在进行一些临床试验,为NP-C患者开发新的治疗选择。
本文由中国罕见病网编译
ID:Niemann_Pick(中文名“NPD公益组织”)

哪里会有人喜欢孤独,只是不愿意失望罢了。——渡边彻《挪威的森林》
2018年5月22日,国家卫生健康委员会、科技部、工信部、国家药监局、国家中医药管理局等五部委联合制定的《第一批罕见病目录》正式发布,共涉及121种罕见病。
目前世界上已知约7000多种罕见病,虽然其发病率极低(0.65‰-1‰),但患者人数不少(已超3亿),而且罕见病一旦发病病情恶化迅速,死亡率高。然而,95%的罕见病目前并没有特效治疗药物,这种无药可治的状态亟待改变。
此次国家5部委联合制定的首批罕见病目录重磅出台,这对加强我国罕见病管理和提高我国罕见病诊疗水平具有非常重要的意义,罕见病行业将迎来新的发展机遇。我们认为后续将有政策从研发、审评、产业化、定价及医保支付多方面对罕见病有关药品及医疗器械给予支持,有望激发国内药企进行罕见病研发投入的热情。
1
21-羟化酶缺乏症
严重时威胁生命。发病率在爱斯基摩人约为1/500,白种人和黄种人为1/2万~1/5万。
辅助检查
1.单纯男性化型:血清17-OHP、雄烯二酮和睾酮水平升高,24h尿17-酮类固醇(17-KS)和17-生酮类固醇(17-KGS)排量增高,血清电解质和醛固酮水平正常,PRA正常或轻度升高。染色体核型正常。
2.失盐型:血清和尿肾上腺皮质类固醇谱和单纯男性化型相同,血浆醛固酮水平降低,PRA水平显著升高,低血钠、低血糖、高血钾、代谢性酸中毒。染色体核型正常。
3.无症状型血清17-OHP、雄烯二酮和睾酮的基础水平或ACTH兴奋的反应水平增高,尿17-KS和17-KGS水平增高。
4.迟发型:与无症状型相似。
单纯男性化型
①女性患者外生殖器有不同程度男性化(从阴蒂肥大到大阴唇融合,形成部分性阴茎尿道)。
②男性患者表现为非促性腺激素释放激素(GnRH)依赖性性早熟,阴茎增大,阴毛生长,但是睾丸和促性腺激素仍停留在青春期前水平。皮肤色素沉着和身体直线生长加速与女性患者表现相同。
失盐型
皮质醇和醛固酮两种合成途径的CYP21都受累。新生儿患者除了具有单纯男性化型的临床表现外,还有水盐失衡的表现;如拒乳、呕吐、脱水、休克。如得不到及时救治,死亡率甚高。失盐型患者大多数在出生后1~5周发病,6周以后发病者极少。
非经典型
①无症状型:又称隐性CYP21缺乏症。在经典型CYP21缺乏症家系中,有些成员无男性化表现,但是血清CYP21催化反应步骤的前体类固醇17-OHP水平增高。
②迟发型:出生时外生殖器正常,没有男性化改变。而在青春期前出现多毛、痤疮、身体直线生长加速和骨龄提前。
2
白化病
所属科室:皮肤科常见病因:近亲结婚
检查
1.基因检查、肿瘤标记物检查。
2.组织病理检查。
临床表现
白化病全身皮肤呈乳白或粉红色,毛发为淡白或淡黄色。由于缺乏黑色素的保护,患者皮肤对光线高度敏感,日晒后易发生晒斑和各种光感性皮炎。并可发生基底细胞癌或鳞状细胞癌。眼部由于色素缺乏,虹膜为粉红或淡蓝色,常有畏光、流泪、眼球震颤及散光等症状。
3
Alport综合征(遗传性肾炎)
所属科室:内科-肾内科主要病因:基因突变
发病原因
IV型胶原6条ɑ链聚合成3条三股螺旋分子结构称为单体,单体聚合形成二聚体或四聚体,再相互绞连形成胶原网状结构。X连锁显性遗传Alport综合征得基因突变主要发生在编码IV型胶原ɑ5链的基因(COL4A5)。常染色体隐性遗传Alport综合征的基因突变则发生在2号染色体上编码IV型胶原ɑ3链或者ɑ4链的基因(COL4A3/COL4A4)上。
主要症状:血尿,肾功能进行性减退,耳聋和眼部异常
诊断:Alport综合征主要依据临床表现、家族史、组织基膜IV胶原ɑ链免疫荧光学检查、肾组织活检电镜以及基因分析
4
肌萎缩侧索硬化
所属科室:神经科、皮肤科多发人群:40-50岁男性
常见病因
不明,可能与遗传及基因缺陷有关
常见症状
早期仅无力、肉跳、易疲劳,可吞咽,讲话困难
检查
广州供卵助孕机构要早期诊断肌萎缩侧索硬化,除了神经科临床检查外,还需做肌电图、神经传导速度检测、血清特殊抗体检查、腰穿脑脊液检查、影像学检查,甚至肌肉活检。
5
Angelman氏症候群(天使综合征)
主要病因:基因缺陷,15号染色体q11-q13缺失所致
多发群体:儿童
检查方法:脑电图扫描(EEG)
主要症状:脸上常有笑容,智能低下,缺乏语言能力、过动,癫痫等症状
治疗方法:唤醒沉睡的正常的等位基因
6
精氨酸酶缺乏症未有详细资料
7
热纳综合征(窒息性胸腔失养症)未有详细资料
8
非典型溶血性尿毒症
临床病因:补体调控蛋白H因子、以及膜辅助蛋白和血清补体固有成分(B因子、补体C3)的基因突变都可参与其发病
临床表现:
最主要特点有三:微血管溶血性贫血、血小板减少及肾功能衰竭。
治疗
传统上可供选择的治疗aHUS患者的方式仅限于支持治疗:
·血浆置换/输注治疗:须于诊断后24小时内立即施行,但有些特殊情况的病童可能因病情发展而无法使用血浆置换治疗,而长期使用血浆置换治疗的患者,也可能出现疗效受限的情形。但此治疗对于MCPmutation的病患并无治疗效果。
·免疫抑制剂治疗,但对此类病患效果有限,除了CFHautoantibodies的病患,可以合并血浆置换疗法进行治疗。
·肾功能衰竭患者接受透析治疗
·肾功能衰竭患者接受肾脏移植治疗
·肝-肾联合移植

我不知道世间有什么是确定不变的,我只知道,只要一看到星星,我就会开始做梦。——文森特·梵高
9
自身免疫性脑炎未有详细资料
11
自身免疫性胰岛素受体病未有详细资料
12
β-酮硫解酶缺乏症
多发群体
5个月大至2岁之间
常见病因
由于肠胃炎和发热性疾病,如上呼吸道感染,麻疹,或中耳炎等症状而被诱发
常见症状
呕吐,脱水,呼吸急促或呼吸困难,肌张力低下,嗜睡有时候会导致昏迷
检查:可经由尿液有机酸分析和血片串联质谱仪分析来判断个案是否患有β-酮硫解酶缺乏症
皮肤切片进行酵素活性检测及基因检测亦可作为确认诊断的方法之一
12
生物素酶缺乏症未有详细资料
13
心脏离子通道病
就诊科室:心血管内科
常见发病:心脏
常见病因:多数源于编码离子通道亚基单位的基因突变
常见症状:平时无心绞痛发作、胸闷、呼吸困难等,通常以晕厥或猝死为首发表现,其特点为发作前无先兆症状,多发生在夜间睡眠状态,伴呻吟、呼吸浅慢、呼吸困难。
检查
1.实验室检查
主要是血清离子测定,可有电解质紊乱表现。
2.心电图
长Q-T间期综合征可表现为尖端扭转型室性心动过速;短Q-T间期综合征表现为ST段近乎消失,常伴T波高尖、窄且不对称,Q-T间期300ms;布鲁加达综合征典型表现为右胸导联ST段呈下斜型或马鞍型抬高。
治疗
1.一般治疗
戒除不良生活习惯等。
2.药物治疗
应用β受体拮抗药及相应离子通道阻滞药对某些离子通道病可能有效,药物通过不同机制抑制室性心动过速、心室颤动的诱发,可作为本病的辅助治疗措施;另外,补充相应的离子也是本病有效的辅助治疗方法。
3.手术治疗
植入型心律转复除颤器对大多数心脏离子通道病有效,不适合植入除颤器者采用射频消融也有一定作用。
14
原发性肉碱缺乏症
发病部位:细胞线粒
主要症状:发展迟缓,中枢神经功能失常,肝肿大,肝脏脂肪变性
传染性:无传染性
是否进入医保:否
遗传模式:染色体隐性遗传
诊断:在实验室检验项目上,可检验患者血液及尿液中的肉碱浓度、血氨、血糖、尿酮、肝功能、电解质、尿酸、CK(肌肝酸)值、乳酸及凝血功能等数值。并可经心脏超音波、X光等影像学检查,以了解是否发生心脏肥大的情形。在疾病确诊上,为经皮肤切片,以检测细胞中的酵素活性,或抽血经分子生物技术进行缺陷基因的检测。目前国内将「TandemMass串联质谱仪」的技术应用于「新生儿筛检」上,将可能因此可以早期检测出患者,以于早期提供治疗,预防脑部及心脏等器官发生病变。
治疗
在治疗上,平时可口服补充肉碱,并避免饥饿及禁食的情形发生,以防症状的恶化。当急性发作,而出现低酮性低血糖的脑神经病变时,则应先给予患者葡萄糖静脉输液;若已确认为此症患者,则可经由静脉补充肉碱,以提升组织内的肉碱含量,将有利于粒线体内的脂肪酸运输及代谢。长期治疗的患者,可以口服的方式补充肉碱。在过去的治疗经验上,我们曾在一名需要接受换心手术的孩童身上,诊断他为此症;在给予肉碱补充,使得心脏功能得以改善,因此免除了他接受换心手术。
15
Castleman病
实验室检查
1.外周血轻至中度正细胞正色素性贫血,部分病例有白细胞和(或)血小板减少也可表现为典型火罐网的慢性病性贫血。
2.骨髓象部分患者浆细胞升高自2%~20%不等,形态基本正常。
3.血液生化及免疫学检查肝功能可异常,表现为血清转氨酶及胆红素水平升高少数病人肾功受累血清肌酐水平上升血清免疫球蛋白呈多克隆升高,较为常见,少数血清出现M蛋白,血沉也相应增快。部分患者抗核抗体类风湿因子及抗人球蛋白试验阳性。
4.尿常规尿蛋白轻度升高,如伴发肾病综合征,则出现大量蛋白。
其它检查:
根据临床表现、症状、体征选择病理检查、X线、CT、B超及心电图等检查。
相关检查:
Coombs试验
单克隆丙种球蛋白
抗核抗体
浆细胞
类风湿因子
肌氨酸酐
蛋白定量(尿)
血小板
血沉
辅助检查
1.组织病理学肿大的淋巴结活检显示上述CD的特殊病理改变。病变主要累及身体任何部位的淋巴组织,偶可波及结外组织CD病理上分为以下两种类型:
(1)透明血管型:占80%~90%淋巴结内显示许多增大的淋巴滤泡样结构,呈散在分布。有数根小血管穿入滤泡,血管内皮明显肿胀,管壁增厚,后期呈玻璃样改变。血管周围有数量不一的嗜酸性或透明状物质分布。滤泡周围由多层环心排列的淋巴细胞,形成特殊的洋葱皮样结构或帽状带滤泡间有较多管壁增厚的毛细血管及淋巴细胞、浆细胞、免疫母细胞,淋巴窦消失,或呈纤维化。大体标本见淋巴结直径为3~7cm,大者可达25cm,重量达700g
(2)浆细胞型:占10%~20%。淋巴结内也显示滤泡性增生但小血管穿入及滤泡周围的淋巴细胞增生远不及透明血管型明显,一般无典型的洋葱皮样结构。本型的主要特征为滤泡间各级浆细胞成片增生,可见Russell小体,同时仍有少量淋巴细胞及免疫母细胞。有人称本型为透明血管型的活动期,可有TCRβ或IgH基因重排。少数患者病变累及多部位淋巴结,并伴结外多器官侵犯,病理上同时有上述两型的特点称为混合型。也有少数单一病灶者病理上兼有上述二型的特点,则为另一意义上的混合型。有报道少数浆细胞型患者可并发卡波西肉瘤,以艾滋病伴发CD者多见。
2.根据临床表现、症状、体征选择X线、CT、B超及心电图等检查。
治疗
局灶型CD均应手术切除,绝大多数患者可长期存活,复发者少。病理上为浆细胞型的局灶性CD,如伴发全身症状,在病变的淋巴结切除后也可迅速消失。
多中心型CD,如病变仅侵及少数几个部位者,也可手术切除,术后加用化疗或放疗病变广泛的多中心型CD只能选择化疗,或主要病变部位再加局部放疗,大多仅能获部分缓解。化疗通常选用治疗恶性淋巴瘤的联合化疗方案。自体造血干细胞移植也是一种治疗选择。
16
腓骨肌萎缩症
检查:神经电生理检查
本病主要分为两型
I型(脱髓鞘型)
是典型的腓骨肌萎缩症双下肢呈倒立酒瓶状或称鹤立腿同时出现足弓高耸爪形趾马蹄内翻畸形等行走时表现特殊的跨越步态表现肌无力肌萎缩肌束颤腱反射减退或消失首发于手部肌前臂肌萎缩而后下肢远端肌萎缩者仅见于少数病例四肢末梢可出现手套状袜状深浅感觉障碍和一系列植物神经与营养代谢障碍局部皮肤呈青紫色皮肤温度低溃疡形成等
II型(轴突型)
发病晚成年开始有肌萎缩部位和症状与I型相似但程度较I型为轻临床上按症状组合有多种变异型如肩胛腓骨肌萎缩型视神经萎缩型弗里德赖希共济失调若伴有腓骨肌萎缩则称腓骨肌萎缩型共济失调即Roussy-Levy综合征
腓骨肌萎缩症的治疗
本病目前尚未无特殊有效治疗,主要是对症和支持疗法,垂足和足畸形可穿着矫鞋,由于病程进展缓慢,大多数患者可存活数十年,对证治疗可提高患者生活质量。
26
瓜胺酸血症
诊断方法
包括检验血中血氨值,血中瓜胺酸值,及其他相关血中胺基酸与肝功能检查。目前可做基因检查来确认诊断,约有80%的个案可以找到基因突变点。
先天性高胰岛素行低血糖血症未有详细资料
先天性肌强直(非营养不良性肌强直综合征)
就诊科室
骨科、神经内科
常见病因
遗传
常见症状
肌强直和肌肥大
常见发病
肢体近端肌肉、眼睑、舌
检查方法:肌电图检查、心电图检查、产前检查、血尿常规检查、
特殊实验:(1)冷水诱发实验将手及前臂浸入11~13℃冷水数分钟至40分钟,可诱发肌强直及肌无力;
(2)钾负荷试验让疑诊者口服氯化钾后,看能否诱发明显的肌无力。钾盐诱发的无力一般在1小时内达高峰,1.5小时恢复正常,钾盐应从小量开始,避免心律失常。
治疗
1.本病目前尚无根治的方法,只能针对其肌强直症状,进行对症治疗。由于相当一部分轻症患者已能适应肌强直状态,对日常生活影响不大,因此不需特殊治疗。对肌强直严重的病例,已影响生活和工作时可给予药物治疗;常显遗传病人在病程中通常无明显加重。常隐遗传者可缓慢加重,并可伴有一过性肌无力;
2.先天性肌强直与强直性肌营养不良的治疗原则相同,对局麻药、抗心律失常药反应较好,这类药物主要对钠通道起抑制作用,对氯通道的作用尚不清楚。美西律是首选药物。
3.先天性新生儿肌强直者需注意喂养,避免吸入性肺炎。副肌强直症者应注意保暖,避免在寒冷环境工作。服药过程中应注意药物的副反应,需定期检查血象、心电图等。乙酰唑胺、卡马西平、氯硝西泮也有一定疗效,中等剂量皮质激素可减轻肌强直。
18
先天性脊柱侧弯
就诊科室骨科
常见发病脊柱
常见病因先天性椎体异常,病因不清
临床表现
早期轻型脊柱侧凸的征象
1.两肩不等高;
2.肩甲一高一低;
3.一侧腰部皱褶皮纹;
4.腰前屈时两侧背部不对称,即“剃刀背”;
5.脊柱偏离中线。
检查
检查时将幼儿从腋下悬吊起观察侧凸的僵硬度、可屈性,进行神经系统检查,有无肌张力增高或低下,了解有无其他先天性畸形,拍悬吊位及仰卧位脊柱全长正侧位片,观测肋椎角差异。明显的脊柱侧凸一般体格检查即可确定诊断,但是对于侧凸的角度,仍需要经X线摄片方能最后确定。
治疗:1.非手术治疗
先天性脊柱侧凸最常用的非手术治疗是观察,仅适用于自然史不清楚的病例,在半椎体或混合畸形中观察可起到一定作用,而对于一侧骨桥形成的患者则不适宜观察。观察方法:每4~6个月随诊一次,常规行站立位脊柱全长正侧位X线检查,对不能站立的婴幼儿可行卧位X线检查。
支具疗法适应证是有一定限度的,对尚未发育成熟、畸形逐渐加重,侧弯节段长且柔软的患者适应于支具治疗。对无进展的病例不需要应用支具,更不适用于畸形已有自动改善的病例。而对节段短且僵硬的病例,支具治疗几乎无效。
2.手术治疗
(1)后路原位融合术指不应用器械矫形的后路、脊柱融合,适用于孤立或短节段的单侧骨桥或半椎板典型畸形出现之前,年龄5岁的幼儿。
(2)凸侧骨髓阻滞术适用于单侧形成不良的患者,如完全分解的半椎体畸形,通过对凸侧骨髓及生长终板的抑制,保留凹侧的生长潜能。
(3)后路器械矫形融合术常用于年龄较大或畸形严重的先天性脊柱侧凸。
(4)半椎体切除术可以直接去除致畸因素,可以分为前后路联合一期或二期半椎体切除术和后路半椎体切除术两种。
(5)脊柱截骨术通常应用于骨盆倾斜、躯干失代偿、进展性畸形及脊髓神经异常的患者,联合半椎体切除及器械矫形融合可对严重的畸形加以矫正。
(6)生长棒技术是针对早发进展性脊柱侧凸,分为单棒技术和双棒技术。
(7)胸廓扩大成形术和垂直挣开扩展胸廓钛肋骨假体术适用于有椎体畸形并伴有肋骨融合的早期先天性畸形,通过胸壁切开或并肋切断,置入可持续撑开的人工肋骨,使胸廓畸形得到矫正。

冠状动脉扩张病未有详细资料
先天性纯红细胞再生障碍性贫血
Erdheim-Chester病
法布雷病
家族性地中海热
范可尼贫血
半乳糖血症
戈谢病
全身型重症肌无力
Gitelman综合征
戊二酸血症I型
糖原累积病(I型II型)
血友病
肝豆状核变性
遗传性血管性水肿
遗传性大疱性表皮松解症
遗传性果糖不耐受症
遗传性低镁血症
遗传性多发性脑梗死性痴呆
遗传性痉挛性截瘫
全羧化酶合成酶缺乏症
同型半胱氨酸血症
纯合子家族性高胆固醇血症
亨廷顿舞蹈病
HHH综合征
高苯丙氨酸血症
低碱性磷酸酶血症
低磷性佝偻病
特发性心肌病
特发性低促性腺激素性腺功能减退症
特发性肺动脉高压
特发性肺纤维化
IgG4相关性疾病
先天性胆汁酸合成障碍
异戊酸血症
卡尔曼综合征
朗格汉斯组织细胞增生症
莱伦氏综合征
Leber遗传性视神经病变
长链3-痉酰基辅酶A脱氢酶缺乏症
淋巴管肌瘤病
赖氨酸尿蛋白不耐受症
溶酶体酸性脂肪酶缺乏症
枫糖尿病
马凡综合征
McCune-Albrigh综合征
中链酰基辅酶A脱氢酶缺乏症
甲基丙二酸血症
线粒体脑肌病
黏多糖贮积病
多灶性运动神经病
多种酰基辅酶A脱氢酶缺乏症
多发性硬化
多系统萎缩
肌强直性营养不良
N-乙酰谷氨酸合成酶缺乏症
新生儿糖尿病
视神经脊髓炎
尼曼匹克病
非综合征性耳聋
Noonan综合征
鸟氨酸氨甲酰基转氨酶缺乏症
成骨不全症(脆骨病)
帕金森病(青年型、早发型)
阵发性睡眠性血红蛋白尿
黑斑息肉综合征
苯丙酮尿症
POEMS综合征
原发性联合免疫缺陷
原发性遗传性肌张力不全
原发性轻链型淀粉样变
进行性家族性肝内胆汁淤积症
进行性肌营养不良
丙酸血症
肺泡蛋白沉积症
肺囊性纤维化
视网膜色素变性症
视网膜母细胞瘤
重症先天性粒细胞缺乏症
婴儿严重肌阵挛性癫痫(Dravet综合征)
镰刀型细胞贫血症
Silver-Russell综合征
谷固醇血症
脊髓延髓肌萎缩症(肯尼迪病)
脊髓型肌萎缩症
脊髓小脑性共济失调
系统性硬化症
四氢生物蝶呤缺乏症
结节性硬化症
原发性络氨酸血症
极长链酰基辅酶A脱氢酶缺乏症
威廉姆斯综合征
湿疹血小板减少伴免疫缺陷综合征
X-连锁无丙种球蛋白血症
X-连锁肾上腺脑白质营养不良
X-连锁淋巴增生症
他是你前世遗落的一滴泪,今生,不管谁人装饰你的梦,他的梦只有你是唯一的点缀;不管你呼唤的人是谁,你的名字是他心里梦里唯一的执念不悔。
欢迎分享,本文由医百病ID:VIPTMi整理发布
广州德清供卵公司,广州供卵哪里有,广州代孕2万起,广州专业供卵价格表,广州全国供卵中心,广州国内是不是不能供卵,广州弱精症做试管几率,广州高龄供卵试管,广州男人冻精子,广州怎样防止供卵自怀受骗,